Photodynamic therapy (PDT) has emerged as a powerful approach in precision medicine. This technique offers highly targeted treatment, allowing doctors to activate therapy exactly where and when it is needed while minimizing damage to surrounding healthy tissue. Because it is non-invasive and associated with relatively few side effects, PDT is increasingly being explored for the treatment of certain cancers, benign lesions, and even infectious diseases.
The principle behind PDT is simple and straightforward. A light-sensitive compound, a photosensitizer molecule, is introduced into the body and accumulates preferentially in diseased cells. When this molecule is exposed to a specific laser-light irradiation and oxygen is present in the tissue, it triggers a chemical reaction that generates cytotoxic reactive oxygen species (ROS).
These compounds selectively damage and destroy the targeted cells. In this process, light, oxygen, and photosensitizer are the three essential components of PDT, with the photosensitizer being the key factor determining therapeutic efficacy.
Challenges in Photosensitizer Design and Optimization
However, the discovery of high-performance photosensitizers has long been hindered by the time-consuming and resource-intensive nature of traditional trial-and-error approaches.
Designing effective photosensitizers requires simultaneously optimizing multiple competing properties, high singlet oxygen quantum yield (ϕΔ) for therapeutic potency and long absorption wavelength (λmax) for deep tissue penetration, a balance that traditional chemistry struggles to achieve.
The relevant chemical space is vast, and experimental screening is slow and expensive. While density functional theory (DFT) and time-dependent DFT (TD-DFT) can compute ground-state and excited-state properties, their computational cost scales as O(N³) or higher with system size, making large-scale rapid screening impractical.
Recent machine learning approaches in photosensitizer design have focused mainly on predicting properties of known molecules, but no AI-generated photosensitizer had previously undergone experimental validation demonstrating clinically relevant performance.
A critical bottleneck has been the absence of comprehensive excited-state property databases needed to train domain-specific AI models, particularly for experiment-derived photochemical properties.
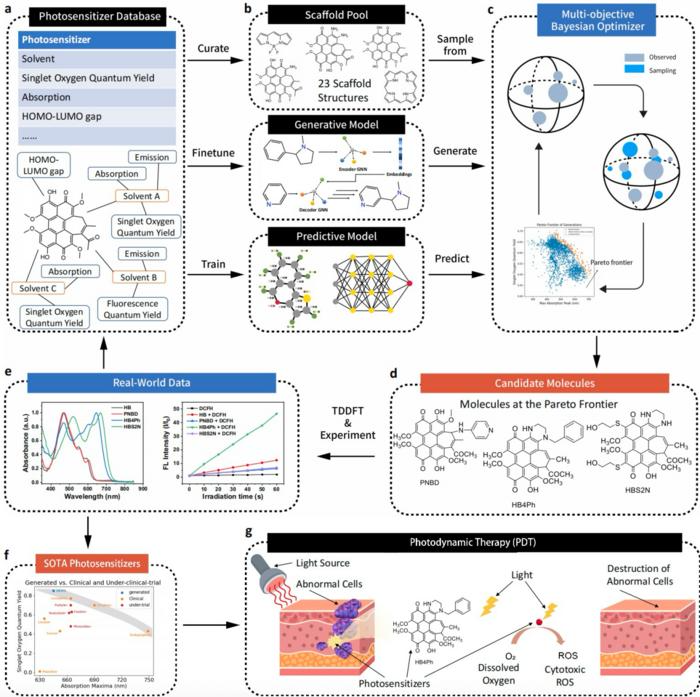
AAPSI Workflow for AI-Driven Molecular Discovery
The AAPSI workflow addresses these challenges through an integrated pipeline spanning data curation, AI-driven molecular design, and experimental validation.
The workflow of AAPSI, the AI-accelerated workflow for photosensitizer discovery. Image Credit: Hongyi Wang from City University of Hong Kong, Xiuli Zheng from Technical Institute of Physics and Chemistry, and Sheng Gong from Massachusetts Institute of Technology
Large-Scale Photosensitizer Database Creation
The team created a database of 102,534 photosensitizer-solvent pairs encompassing 23,650 unique molecules. The largest collection of its kind. Organized into six subsets covering absorption maxima, emission maxima, HOMO-LUMO gaps, singlet oxygen quantum yield, fluorescence quantum yield, and a complete molecular repository, the database captures the critical influence of solvent environment on photochemical properties. It spans diverse structural classes, including porphyrins, BODIPYs, phenothiazines, xanthenes, cyanines, phthalocyanines, and perylenequinones. The database is publicly accessible at http://aapsi.online.
AI Models for Molecular Generation and Screening
AAPSI employs two complementary AI models. SolutionNet, a graph transformer with dual-input architecture, predicts ϕΔ and λmax with uncertainty quantification by jointly processing photosensitizer and solvent molecular graphs. MoLeR, a scaffold-based generative model built on an encoder-decoder framework, produces novel molecules while preserving core photoactive scaffolds to ensure synthetic feasibility.
The team curated 23 scaffolds derived from natural products - including hypocrellin, elsinochrome, hypericin, porphyrins, and BODIPYs - embedding expert knowledge into the generative process.
Two-generation strategies yielded 6,148 unique candidates: direct scaffold-based generation (3,660 molecules) and multi-objective Bayesian optimization using the qNEHVI acquisition function with Matérn 5/2 Gaussian process kernels (2,488 molecules). The MOBO-generated molecules progressively expanded the Pareto frontier of simultaneously high ϕΔ and long λmax. After screening for synthetic accessibility and drug-likeness, 9 candidates at the Pareto frontier were selected for further investigation. Notably, the entire computational pipeline runs on a single consumer-grade GPU (NVIDIA RTX 4090).
Experimental Validation of AI-Designed Molecules
From the 9 Pareto-optimal candidates, three hypocrellin derivatives (HB4Ph, PNBD, and HBS2N) were selected for synthesis and comprehensive experimental characterization, including TD-DFT pre-validation, absorption and fluorescence spectroscopy, ROS generation assays, and singlet oxygen quantum yield measurements. Hypocrellin derivatives were prioritized because hypocrellin is a natural product-derived scaffold with proven photodynamic potential and is regarded as a next-generation photosensitizer for PDT.
HB4Ph emerged as the standout candidate: its absorption maximum at 645 nm and emission at 713 nm approach the near-infrared therapeutic window (700–1200 nm) ideal for deep-tissue tumor treatment, while its singlet oxygen quantum yield of 0.85 surpasses all clinical and trial-stage photosensitizers. Compared to the parent molecule hypocrellin B (HB; ϕΔ = 0.76, λmax = 467 nm), HB4Ph achieves dramatic improvements in both key metrics.
The weak fluorescence of HB4Ph indicates efficient intersystem crossing (ISC) to the triplet state, consistent with its exceptionally high ϕΔ. Mechanistically, the incorporation of two nitrogen atoms into the conjugated π-system enhances spin-orbit coupling, promoting ISC and subsequent singlet oxygen generation. HB4Ph sits on the Pareto frontier among all clinical and trial-stage organic molecular photosensitizers for PDT, optimally balancing therapeutic potency with tissue penetration depth.
The other two candidates provided complementary insights: HBS2N, incorporating two nitrogen and two sulfur atoms, achieved the longest absorption wavelength (668 nm) but the lowest ϕΔ (0.10), revealing a trade-off between spectral red-shifting and photodynamic efficiency. PNBD, with a pyridine group in non-conjugated regions, showed performance similar to HB (λmax = 472 nm, ϕΔ = 0.73), confirming that conjugation-site modification is key to property enhancement.
Future Directions for Multi Objective Optimization
While AAPSI currently optimizes two key properties, the framework is designed for extensibility. Future development will incorporate additional objectives such as water solubility, biocompatibility, and pharmacokinetic (ADMET) properties into the multi-objective optimization.
The team plans to establish a dynamic experimental feedback mechanism that enables models to continuously learn from new validation data. Hybrid generation strategies combining scaffold-constrained and free-form molecular design will further expand accessible chemical space. Beyond photosensitizers, the AAPSI framework is readily transferable to other domains requiring multi-property molecular optimization, including drug discovery, catalyst design, and functional materials development.
Impact of AI-Driven Molecular Discovery Pipeline
This work represents a milestone in AI-driven molecular discovery: HB4Ph is the first AI-designed photosensitizer to achieve state-of-the-art performance, as validated experimentally, demonstrating that AI can identify high-performance candidates in chemical spaces that traditional methods have not reached. The publicly available database of 102,534 photosensitizer-solvent pairs fills a critical gap in excited-state property data for the research community.
The AAPSI workflow establishes a complete pipeline from AI molecular design to experimental validation, demonstrating that integrating expert domain knowledge with AI-powered generation and multi-objective optimization can compress the molecular discovery cycle from years to days. All data and code have been made publicly available (database: http://aapsi.online; code: https://github.com/howardwang1997/AI4PS), offering a replicable and extensible paradigm for accelerated materials innovation across scientific disciplines.
Study Citation and Resources
Citation: Hongyi Wang, Xiuli Zheng, Weimin Liu, Zitian Tang, Sheng Gong. Artificial intelligence driven workflow for accelerating design of novel photosensitizers[J]. AI for Science, 2026, 2(1): 015002. DOI: 10.1088/3050-287X/ae4412
Source:
Journal reference:
 How is One Scientist Using Human Cognition to Build Better AI?
How is One Scientist Using Human Cognition to Build Better AI?